本文作者:余光创,目前就读于香港大学公共卫生系,开发过多个R/Bioconductor包,包括 ChIPseeker, clusterProfiler, DOSE, ggtree, GOSemSim 和 ReactomePA。
进化树看起来和层次聚类很像。有必要解释一下两者的一些区别。
层次聚类的侧重点在于分类,把距离近的聚在一起。而进化树的构建可以说也是一个聚类过程,但侧重点在于推测进化关系和进化距离(evolutionary distance)。
层次聚类的输入是距离,比如euclidean或manhattan距离。把距离近的聚在一起。而进化树推断是从生物序列(DNA或氨基酸)的比对开始。最简单的方法是计算一下序列中不匹配的数目,称之为hamming distance(通常用序列长度做归一化),使用距离当然也可以应用层次聚类的方法。进化树的构建最简单的方法是非加权配对平均法(Unweighted Pair Group Method with Arithmetic Mean, UPGMA),这其实是使用average linkage的层次聚类。这种方法在进化树推断上现在基本没人用。更为常用的是邻接法(neighbor joining),两个节点距离其它节点都比较远,而这两个节点又比较近,它们就是neighbor,可以看出neighbor不一定是距离最近的两个节点。真正做进化的人,这个方法也基本不用。现在主流的方法是最大似然法(Maximum likelihood, ML),通过进化模型(evolutionary model)估计拓朴结构和分支长度,估计的结果具有最高的概率能够产生观测数据(多序列比对)。另外还有最大简约法和贝叶斯推断等方法用于构建进化树。
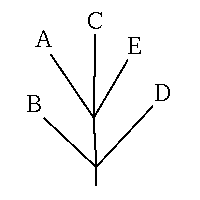
是最常用的存储进化树的文件格式,如上面这个树,拓朴结构用newick格式可以表示为:
(B,(A,C,E),D);
括号最外层是根节点,它有三个子节点,B, (A,C,E)和D,而节点(A,C,E)也有三个子节点A,C和E。
加上分支长度,使用:来分隔:
(B:6.0,(A:5.0,C:3.0,E:4.0):5.0,D:11.0);
比如A:5.0代表的是A与其父节点的距离是5.0。
内部节点也可以有label,写在相应的括号外面,如下所示:
(B:6.0,(A:5.0,C:3.0,E:4.0)Ancestor1:5.0,D:11.0);
这是最为广泛支持的文件格式,很多进化树可视软件只支持newick格式。
ggtree的开发源自于我需要在树上做注释,发现并没有软件可以很容易地实现,通常情况下我们把统计信息加到节点的label上来展示,比如CodeML的dN/dS分析,输出文件里就给用户准备了newick树文本,把dN/dS ( \(\omega\) ) 加于节点label之上:
codeml_file <-system.file("extdata/PAML_Codeml/mlc", package="ggtree")
tree_text <-readLines(codeml_file)[375:376]
tree_text
# [1] "w ratios as labels for TreeView:"
# [2] "(K #0.0224 , N #0.0095 , (D #0.0385 , (L #0.0001 , (J #0.0457 , (G #0.1621 , ((C #0.0461 , (E #0.0641 , O #0.0538 ) #0.0001 ) #0.0395 , (H #0.1028 , (I #0.0001 , (B #0.0001 , (A #0.0646 , (F #0.2980 , M #0.0738 ) #0.0453 ) #0.0863 ) #1.5591 ) #0.0001 ) #0.0001 ) #0.0549 ) #0.0419 ) #0.0001 ) #0.0964 ) #0.0129 );"
这种做法只能展示一元信息,而且修改节点label真心是个脏活,满满的都是不爽,我心中理想的方式是树与注释信息分开,注释信息可以方便地通过图层加上去,而且可以自由组合。于是着手开发ggtree是个简单易用的R包,一行代码
ggtree(read.tree(file))
即可实现树的可视化。而注释通过图层来实现,多个图层可以完成复杂的注释,这得力于ggtree的设计。其中最重要的一点是如何来解析进化树。
ggtree的设计
进化树的解析
除了ggtree之外,我所了解到的其它画树软件在画树的时候都把树当成是线条的集合。很明显画出来的进化树就是在画一堆线条,但是线条表示的是父节点和子节点的关系,除此之外没有任何意义,而节点在进化树上代表物种,叶子节点是我们构建进化树的物种,内部节点是根据叶子节点推断的共同祖先。我们所有的进化分析、推断、实验都是针对节点,节点才是进化树上有意义的实体。这是ggtree设计的基础,ggtree只映射节点到坐标系统中,而线条在 geom_tree 图层中计算并画出来。这是与其它软件最根本的不同,也是ggtree能够简单地用图层加注释信息的基础。
扩展ggplot2
有很多可视化包基于ggplot2实现,包括各种 gg 打头的,号称扩展了ggplot2,支持图形语法(grammar of graphics),我并不认同。虽然基于 ggplot2 产生的图,我们可以用theme来进一步调整细节,用scale_系列函数来调整颜色和标尺的映射,但这些不足以称之为支持图形语法,图形语法最关键核心的部分我认为是图层和映射。
像ggphylo, OutbreakTools和phyloseq这几个包都有基于ggplot2的画树函数,但其实都不支持图形语法,它们所实现的是复杂的函数,画完就完事了,用户并不能使用图层来添加相关的信息。
如果在 OutbreakTools 这个包中:
if (show.tip.label) {
p <- p + geom_text(data = df.tip, aes(x = x, y = y, label = label),
hjust = 0, size = tip.label.size)
}
如果show.tip.label=FALSE,当函数返回p 时 df.tip 就被扔掉,用户想要再加 tip.label 就不可能了。 ggphylo 和 phyloseq 都是类似的实现,这些包把树解析为线条,所以节点相关的信息需要额外的 data.frame 来存储,并且只有极少数的预设参数,比如上面例子中的tip.label。在上面的例子中,用户连更改 tip.label 的颜色都不可能,更别说使用额外的注释信息了。
这几个包所实现的画图函数,都可以很容易地用ggtree实现,并用经过测试,ggtree运行速度比这几个包都要快。更多信息请参考ggtree的wiki页面。
ggtree是真正扩展ggplot2,支持图形语法的包。我们首先扩展ggplot支持tree object做为输入,并实现geom_tree图层来画线条。
library(ggplot2)
library(ggtree)
set.seed(2015-11-26)
tree <-rtree(30)
ggplot(tree, aes(x, y)) + geom_tree()
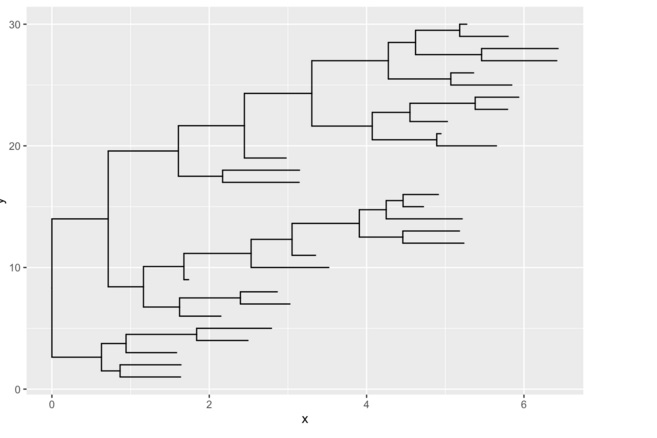
ggtree函数是 ggplot() + geom_tree() + xlab(NA) + ylab(NA) + theme_tree() 的简单组合。
ggtree(tree)
想要加 tip.label,用 geom_tiplab 图层,并且ggplot2的图层都可以直接应用 ggtree。
ggtree(tree) + geom_tiplab() + geom_point(color='firebrick')
 树的操作与注释
树的操作与注释
ggtree提供了多个函数可以把clade放大缩小(scaleClade),折叠(collapse)和展开(expand),位置调换和旋转,以及分类(groupOTU, groupClade)。
nwk <- system.file("extdata", "sample.nwk", package="ggtree")
tree <- read.tree(nwk)
p <- ggtree(tree)
cp <- ggtree(tree) %>% collapse(node=21) + ggtitle('collapse')
ep <- cp + expand(node=21) + ggtitle('expand')
hp <- p + hilight(node=21) + ggtitle('hilight')
rp <- hp + rotate(node=21) + ggtitle('rotate')
library(gridExtra)
grid.arrange(cp, ep, hp, rp, ncol=2)
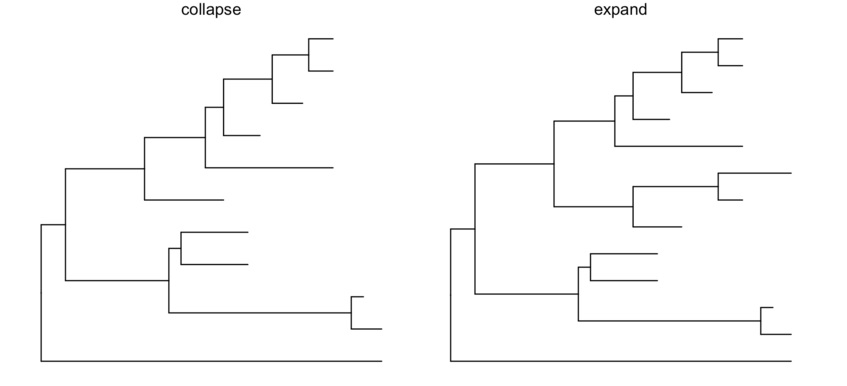
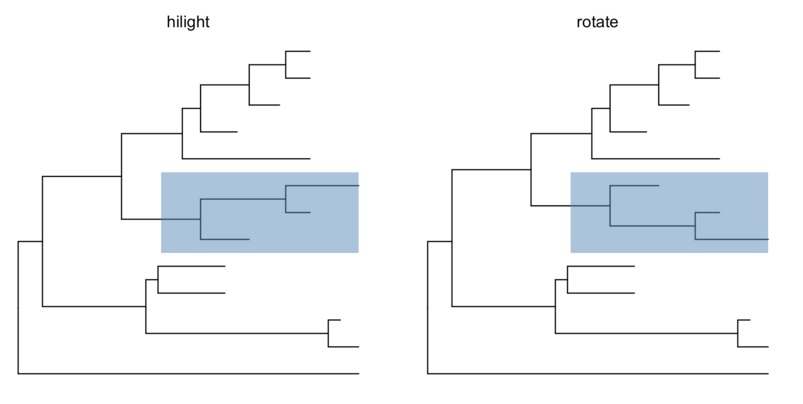
支持多种文件格式
ggtree支持的文件格式包括Newick, Nexus, NHX和jplace。
上面已经展示了Newick格式,下面的例子是NHX格式:
nhxfile = system.file("extdata", "ADH.nhx", package="ggtree")
nhx <-read.nhx(nhxfile)
ggtree(nhx, ladderize=F) + geom_tiplab() + geom_point(aes(color=S), size=8, alpha=.3) +
theme(legend.position="right") +
geom_text(aes(label=branch.length, x=branch), vjust=-.5) +
xlim(NA, 0.3)
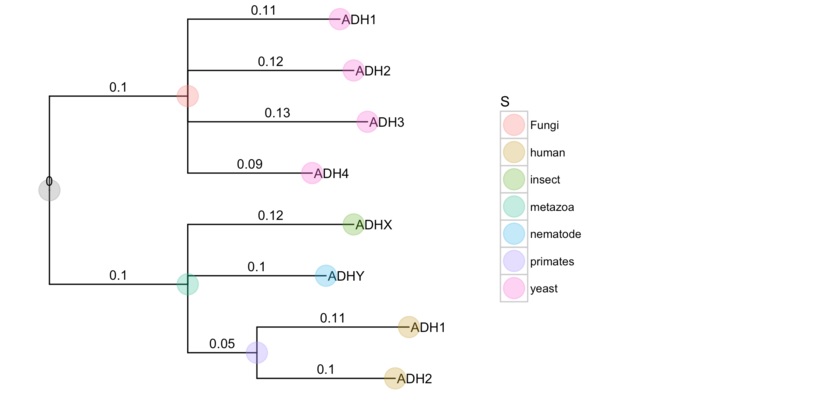 支持解析多种软件的输出文件
支持解析多种软件的输出文件
我们知道FigTree是针对BEAST的输出设计的,可以把BEAST的统计推断拿来给树做注释,但很多的进化分析软件并没有相应的画树软件支持,用户很难把信息展示出来。
ggtree支持ape, phangorn, r8s, RAxML, PAML, HYPHY, EPA, pplacer和BEAST的输出。相应的统计分析结果可以应用于树的注释。可以说ggtree把这些软件分析的结果带给了R用户,通过ggtree的解析, 这些进化分析结果可以进一点在R里进行处理和统计分析,并不单单是在ggtree中展示而已。
RAxML bootstrap分析
raxml_file <-system.file("extdata/RAxML", "RAxML_bipartitionsBranchLabels.H3", package="ggtree")
raxml <-read.raxml(raxml_file)
ggtree(raxml) + geom_text(aes(label=bootstrap, color=bootstrap)) +
scale_color_gradient(high='red', low='darkgreen') +
theme(legend.position='right')
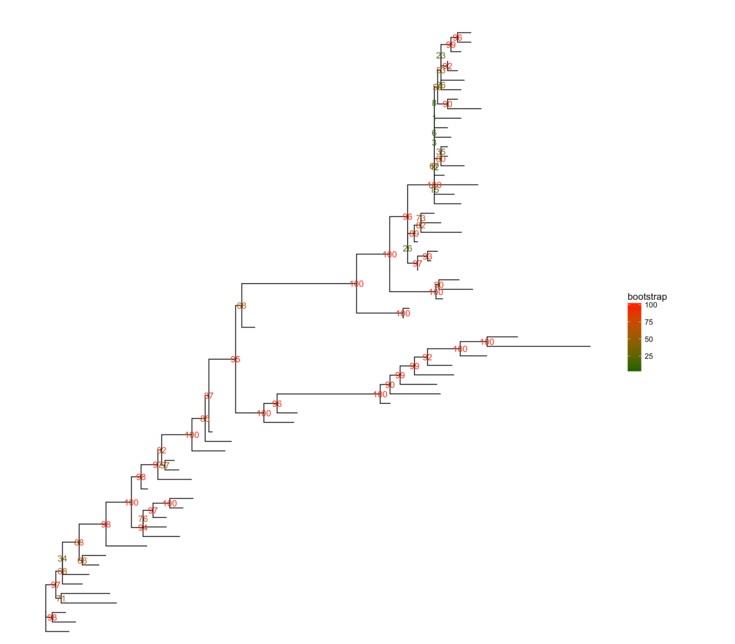
multiPhylo也是支持的,所以100颗bootstrap树可以同时用一行代码展示出来。
btree_file <-system.file("extdata/RAxML", "RAxML_bootstrap.H3", package="ggtree")
btree = read.tree(btree_file)
ggtree(btree) + facet_wrap(~.id, ncol=10)

如果不分面,这100颗树会重叠画在一起,这也能很好地展示bootstrap分析的结果,bootstrap值低的clade,线条会比较乱,而bootstrap值高的地方,线条一致性比较好。
PAML
使用BaseML预测的祖先序列,ggtree解析结果的同时会把父节点到子节点的subsitution给统计出来,可以直接在树上注释:
rstfile <-system.file("extdata/PAML_Baseml", "rst", package="ggtree")
rst <-read.paml_rst(rstfile)
p <-ggtree(rst) + geom_text(aes(label=marginal_AA_subs, x=branch), vjust=-.5)
print(p)
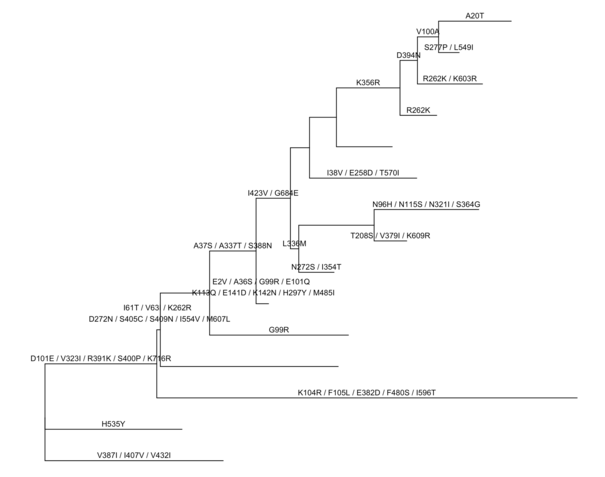
不同于BaseML以碱基为单位,CodeML预测祖先序列,以密码子为单位。`ggtree`定义了一个操作符%<%,如果有相同的注释信息要展示,可以用tree object来更新tree view。
rstfile <-system.file("extdata/PAML_Codeml", "rst", package="ggtree")
crst <-read.paml_rst(rstfile)
p %<% crst
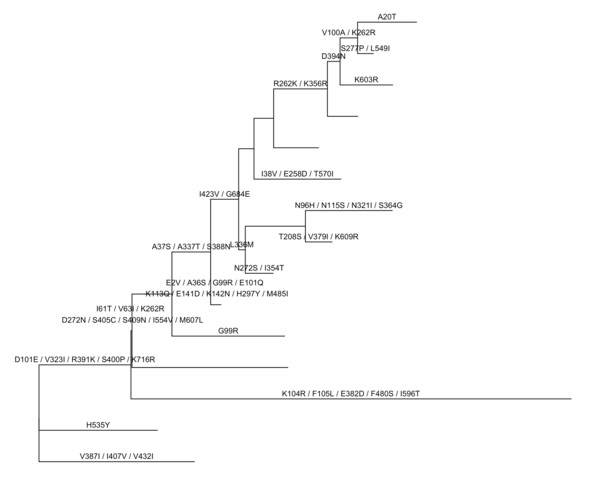
像上面的例子,用crst来更新p,就是用crst画出来的树+注释。对比两图,可以发现BaseML和CodeML推测的祖先序列是稍有不同的。
CodeML的dN/dS分析,我们可以直接把数据拿来给树上色。同样道理分类数据也可以拿来上色。
mlc_file <-system.file("examples/mlc", package="ggtree")
mlc <-read.codeml_mlc(mlc_file)
ggtree(mlc, aes(color=dN_vs_dS)) +
scale_color_continuous(limits=c(0, 1.5), high='red', low='green', oob=scales::squish, name='dN/dS') +
theme(legend.position='right')
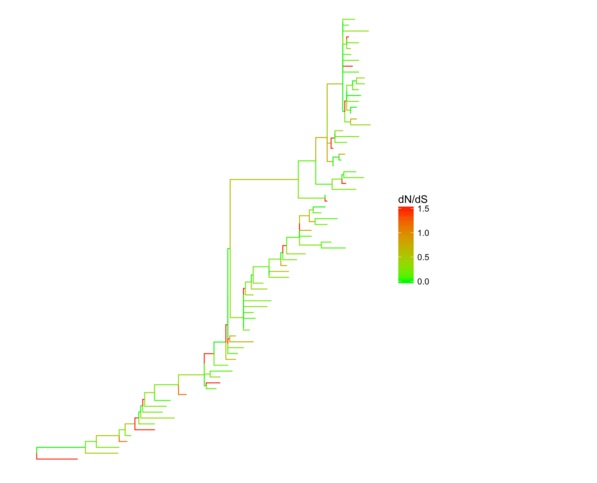 使用用户定义数据
使用用户定义数据
进化树已经被广泛应用于各种跨学科的研究中,随着实验技术的发展,各种数据也更易于获得,使用用户数据注释进化树,也是ggtree所支持的。
nwk <-system.file("extdata", "sample.nwk", package="ggtree")
tree <-read.tree(nwk)
p <-ggtree(tree)
dd <-data.frame(taxa = LETTERS[1:13],
place = c(rep("GZ", 5), rep("HK", 3), rep("CZ", 4), NA),
value = round(abs(rnorm(13, mean=70, sd=10)), digits=1))
## you don't need to order the data
## data was reshuffled just for demonstration
dd <-dd[sample(1:13, 13), ]
row.names(dd) <- NULL
print(dd)

在上面的例子中,使用一个分类数据和一个连续型数据,输入的唯一要求是第一列是taxon label。ggtree中定义了操作符%<+%,来添加数据。添加之后,用户的数据对ggplot是可见的。可以用于树的注释。
p <- p %<+% dd + geom_text(aes(color=place, label=label), hjust=-0.5) +
geom_tippoint(aes(size=value, shape=place, color=place), alpha=0.25)
p+theme(legend.position="right")
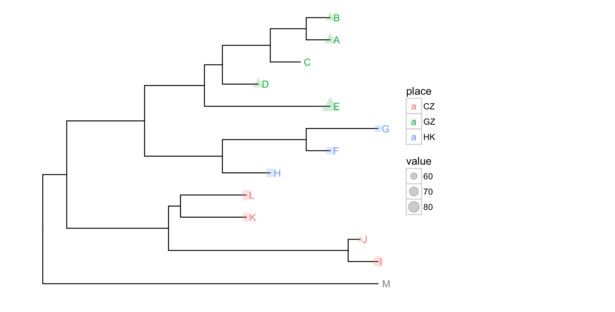
ggtree还支持用户把自己的数据和树保存为jplace格式。
更多的实例请参考vignette。
ggtree允许把不同软件的分析结果整合在一起,同时在树上展示或者比较结果。在我们提交的论文中,使用了整合BEAST和CodeML的例子,在树上展示dN/dS、时间轴、氨基酸替换、clade support values、物种和基因型 (genotype)等多维信息,6种不同的信息同时展示在一颗进化树上,这是个复杂的例子,我们在附件1中展示了可重复的代码。如果有兴趣,可以留意一下我们的文章。 🙂
其它好玩的功能
我们把树当成节点的集合,而不是线条的集合,这一点回归到了进化树的本质意义上,使这一实现成为可能。而支持图形语法,与ggplot2的无缝衔接又让注释变得更加容易ggtree为我们打开了各种注释和操作的可能性。甚至于可以创造出好玩的图,比如使用showtext来加载图形化的字体、 用emoji来画树、使用 图片来注释树等等。
一个比较正经又好玩的是使用PhyloPic数据库上的图形。
pp <-ggtree(tree) %<+% phylopic("79ad5f09-cf21-4c89-8e7d-0c82a00ce728", color="steelblue", alpha = .3)
pp + geom_tiplab(align=T, linetype='dashed', linesize=.5) + geom_tippoint(color='firebrick', size=2)
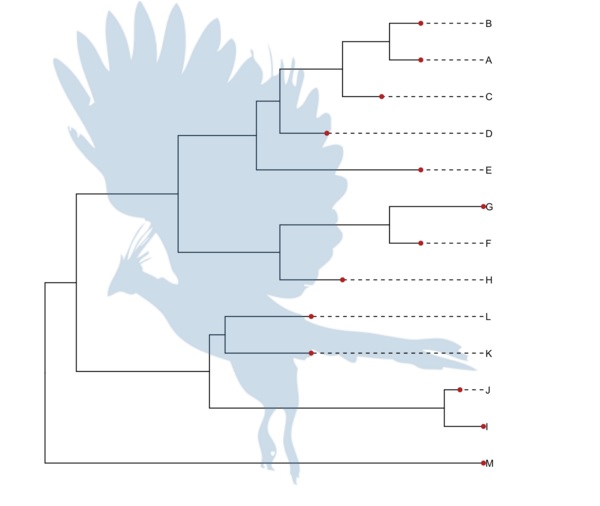
另一个好玩又为我们展现各种可能性的是subview函数,它使得图上加小图变得特别容易。并且已经被应用于地图上加饼图。解决这个问题的初衷在于,想要给节点加饼图注释。有了subview函数之后,这会变得很容易,当然我还没有写出给节点加饼图的函数,因为我还没有这个需求,得有一些实际的数据做参考,这样才能够设计出更易用的函数呈现给用户。
很多的功能还在开发之中,有问题/建议请及时在Github上报告(中英文都可以)。
关于作者
余光创余光创,香港大学公共卫生学院,生物信息学博士生。博客:https://guangchuangyu.github.io, 公众号:biobabble |  |
敬告各位友媒,如需转载,请与统计之都小编联系(直接留言或发至邮箱:editor@cos.name),获准转载的请在显著位置注明作者和出处(转载自:统计之都),并在文章结尾处附上统计之都微信二维码。

发表/查看评论